ABRomics is an online community-driven platform to scale up and improve surveillance and research on antibiotic resistance from a One Health perspective.
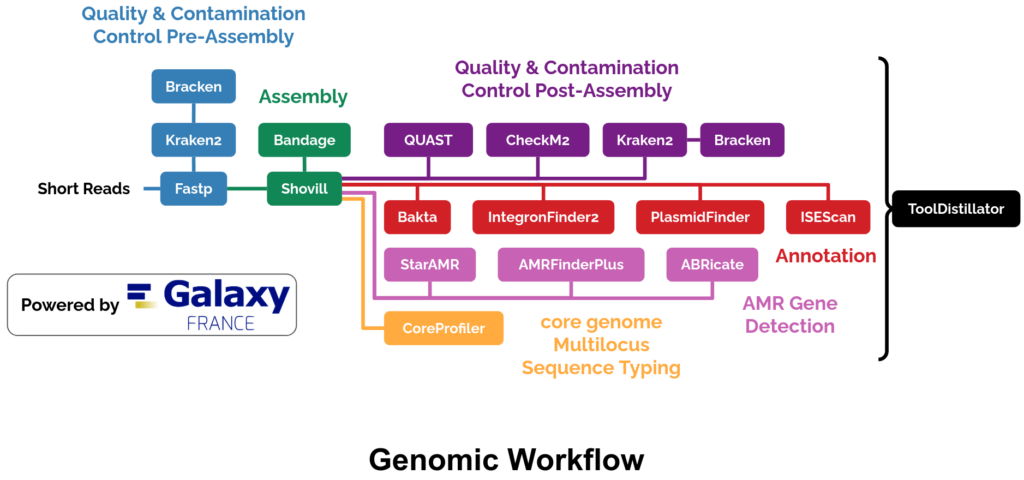
The ABRomics genomic workflow, powered by Galaxy France and launched from the ABRomics platform, is designed to process and analyze bacterial genomic data through a systematic approach. It is divided into six main steps, each ensuring robust and reliable results.
Quality & Contamination Control Pre-Assembly (v1.1.11)
This initial step ensures that raw paired-end Illumina reads are of high quality and control the contamination.
Key Steps:
Quality control and trimming
fastp (Chen et al., 2018) QC control and trimming
Taxonomic assignation on trimmed data
Kraken2 (Wood et al., 2019) assignation
Bracken (Lu et al., 2017) to re-estimate abundance to the species level
Aggregating outputs into a single JSON file
ToolDistillator (ABRomics consortium, 2023) to extract and aggregate information from different tool outputs to JSON parsable files
Outputs:
Quality control:
quality report
trimmed raw reads
Taxonomic assignation:
Tabular report of identified species
Tabular file with assigned read to a taxonomic level
Krona chart to illustrate species diversity of the sample
Aggregating outputs:
JSON file with information about the outputs of fastp, Kraken2, Bracken
core genome Multilocus Sequence Typing (cgMLST) of Bacterial Genome (v1.0)
Performed in parallel with annotation and AMR detection, this step focuses on performing core genome multilocus sequence typing (cgMLST) on contigs.
Key Steps:
Assignment of a cgMLST allele profile on contigs using CoreProfiler (ABRomics consortium, 2023) and curated reference schemes (from pubMLST, BIGSdb, Enterobase, or cgMLST.org)
Aggregating outputs into a single JSON file
ToolDistillator (ABRomics consortium, 2023) to extract and aggregate information from CoreProfiler outputs to JSON parsable files
Outputs:
core genome MLST (cgMLST) results as:
Tabular allele profile for each locus
FASTA file of newly detected alleles
JSON file with information on temporary alleles
Aggregating outputs:
JSON file with information about the outputs of CoreProfiler
The ABRomics workflow provides a comprehensive and integrated approach for bacterial genomic data analysis. From ensuring data quality to identifying critical genes, each step is optimized to deliver actionable and well-organized results.
Useful Links
Galaxy France platform: A web-based platform providing access to powerful, open-source tools for large-scale genomic and metagenomic data analysis.
Bharat A, Petkau A, Avery BP, Chen JC, Folster JP, Carson CA, Kearney A, Nadon C, Mabon P, Thiessen J, Alexander DC, Allen V, El Bailey S, Bekal S, German GJ, Haldane D, Hoang L, Chui L, Minion J, Zahariadis G, Domselaar GV, Reid-Smith RJ, Mulvey MR. Correlation between Phenotypic and In Silico Detection of Antimicrobial Resistance in Salmonella enterica in Canada Using Staramr. Microorganisms. 2022; 10(2):292. 10.3390/microorganisms10020292
Carattoli, A., and H. Hasman, 2020 PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). Horizontal gene transfer: methods and protocols 285–294. 10.1007/978-1-4939-9877-7_20
Chen, S., Y. Zhou, Y. Chen, and J. Gu, 2018 fastp: an ultra-fast all-in-one FASTQ preprocessor. 10.1093/bioinformatics/bty560
Chklovski, A., Parks, D.H., Woodcroft, B.J. et al.CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning.Nat Methods 20, 1203–1212 (2023). https://doi.org/10.1038/s41592-023-01940-w
Feldgarden, M., Brover, V., Gonzalez-Escalona, N. et al.AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence.Sci Rep11, 12728 (2021). 10.1038/s41598-021-91456-0
Gurevich, A., V. Saveliev, N. Vyahhi, and G. Tesler, 2013 QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. 10.1093/bioinformatics/btt086
Lu, J., F. P. Breitwieser, P. Thielen, and S. L. Salzberg, 2017 Bracken: estimating species abundance in metagenomics data. PeerJ Computer Science 3: e104. 10.7717/peerj-cs.104
Néron, B., E. Littner, M. Haudiquet, A. Perrin, J. Cury et al., 2022 IntegronFinder 2.0: identification and analysis of integrons across bacteria, with a focus on antibiotic resistance in Klebsiella. Microorganisms 10: 700. 10.3390/microorganisms10040700
Schwengers, O., L. Jelonek, M. A. Dieckmann, S. Beyvers, J. Blom et al., 2021 Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microbial genomics 7: 000685. 10.1099/mgen.0.000685
Wick, R. R., M. B. Schultz, J. Zobel, and K. E. Holt, 2015 Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31: 3350–3352. 10.1093/bioinformatics/btv383
Wood, D. E., and S. L. Salzberg, 2014 Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biology 15: R46. 10.1186/gb-2014-15-3-r46
Wood, D. E., J. Lu, and B. Langmead, 2019 Improved metagenomic analysis with Kraken 2. Genome biology 20: 1–13. 10.1186/s13059-019-1891-0
Xie, Z., and H. Tang, 2017 ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 33: 3340–3347. 10.1093/bioinformatics/btx433
Zankari, E., H. Hasman, S. Cosentino, M. Vestergaard, S. Rasmussen et al., 2012 Identification of acquired antimicrobial resistance genes. Journal of antimicrobial chemotherapy 67: 2640–2644. 10.1093/jac/dks261